
Болезнь Шарко-Мари-Тута (код по МКБ-10 – G60.0) – это нервно-мышечное заболевание. Термин ШМТ или БШМТ включает не одну болезнь, а группу наследственных расстройств, основными симптомами которых являются слабость мышц конечностей, обычно начинающаяся на ногах (позже также затрагиваются руки), атрофия мышц, нарушение чувствительности.
Что это за болезнь?
Синдром Шарко-Мари-Тута назван в честь 3-х врачей, впервые описавших его в 1886 г. Другой часто используемый термин – наследственная моторно-сенсорная нейропатия (НМСН). Этот термин характеризует 2 основные признаки заболевания:
- наследственность,
- поражение двигательных и сенсорных периферических нервов, приводящее к мышечной атрофии (амиотрофии) и расстройству чувствительности.
Термин «нейропатия» характеризует поражение периферических нервов. БШМТ также часто называют перональной мышечной атрофией (ПМА), т.к. атрофия преимущественно поражает мышцы голени. У пациента с заболеванием важно точно определить форму болезни. Это может быть достигнуто путем тщательного клинического обследования, изучения семейного анамнеза, исследования нервной проводимости и генетического анализа образца крови. Процедура дифференциальной диагностики служит для отличия заболевания от невропатии, не вызванной генетическим дефектом.

Наиболее распространенные виды болезни
Классификация делит БШМТ на несколько типов.
ШМТ 1
Наиболее распространенные типы болезни принадлежат к группе CMT 1 – в первую очередь, это демиелинизирующая полинейропатия с аутосомно-доминантным наследственным типом. Клинически типично раннее начало проявлений в течение 1-2 десятилетий жизни. Биопсия нерва демонстрирует сегментарную демиелинизацию и ремиелинизацию, изображение луковичный формаций (onion bulbs) в сочетании с вторичной потерей аксонов.
ШМТ 1A
В патогенезе применяется дублирование гена периферического белка миелина 22 (PMP 22). При этом типе также были описаны редкие случаи точечных мутаций PMP 22. Тип 1A представляет 60-70% всех нейропатий ШМТ. Типичное начало проявлений приходится на 1-е десятилетие (до 75% пациентов). Типичный признак – нарушение походки, в частности, спотыкание или деформация ноги (высокий подъем).
ШМТ 1B
Это тип первичного демиелинизирующего заболевания Шарко-Мари-Тута с аутосомно-доминантным типом наследования. В качестве причины указывается мутация в гене MPZ (myelin protein zero). Клиническая картина либо сходна с типом 1А, либо является более тяжелой формой ШМТ 1 с ранним началом проявлений.
ШМТ X1
Этот тип составляет около 10% всех случаев болезни и является 2-м наиболее распространенным генетически определенным ее типом. Заболевание вызывается мутациями в гене, кодирующем коннексин 32 (Cx32). X1 – единственный тип болезни, характерный связыванием половой хромосомы X с известной геновой базой. Мужчины болеют раньше и тяжелее, чем женщины. Некоторые женщины длительно остаются без клинических проявлений.
ШМТ 1C
Эта подгруппа была исключена из первичной демиелинизирующей формы, где была продемонстрирована каузальная мутация в гене SIMPLE (small integral membrane protein of lysosome/late endosome). В отличие от равномерного поражения нерва при типе 1A, электрофизиологические признаки 1С включают наличие временной дисперсии и блоков проводимости, главным образом, в области n. Tibialis.
ШМТ 1D
Это редкий тип первичной демиелинизирующей болезни с аутосомно-доминантным типом наследования, где была доказана мутация в гене EGR2 (early growth response). По сравнению с 1A, это более тяжелая форма ШМТ 1 с ранним началом сколиоза. Но также регистрируются случаи с более поздним возрастом появления признаков.
Общие симптомы
Первые симптомы болезни Шарко-Мари-Тута обычно появляются в возрасте 5-15 лет. Иногда признаки начинают проявляться позже, в некоторых случаях даже в среднем возрасте.
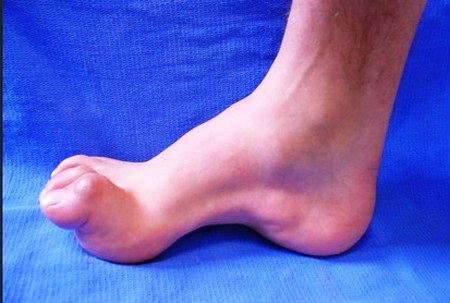
Одно из первых проявлений – ослабление мышц нижних конечностей, характерное для миопатии. Оно приводит к спотыканию, а впоследствии может препятствовать самостоятельной ходьбе. Дальнейшее прогрессирование заболевания сопровождается структурными деформациями ног. Развивается т.н. полая стопа (высокий подъем, укороченное ахиллово сухожилие и молоткообразные пальцы на ногах). Прогрессирующее ослабление мышц приводит к проблемам при ходьбе, беге, поддержке равновесия.
Спотыкание пациенты предотвращают, поднимая колени при ходьбе. У некоторых людей мышечная слабость может возникнуть на уровне бедра.
Вследствие атрофии мышц может нарушиться функция рук, особенно область мелкой моторики (письмо). Невральные поражения приводят к снижению способности различать крошечные, теплые и холодные предметы путем прикосновения.
Хотя болезнь передается наследственно, степень поражения может сильно различаться между пациентами в пределах одной семьи. Ребенок может иметь меньшие поражения, чем его родитель.
Наследование БШМТ
Наследование заболевания в основном аутосомно-доминантное. Это означает, что если у одного из родителей есть болезнь (не имеет значения, у отца или матери), существует вероятность 50% передачи его детям по наследству. Однако наследование также может быть рецессивным или X-связанным. Чтобы определить тип наследственности, проводится специализированное генетическое исследование.
Диагностика
Диагностическая процедура включает ряд методов, среди которых:
- подробный личный и семейный анамнез,
- клиническая оценка мышечной силы, чувствительности,
- электрофизиологическое исследование скорости проводимости нервного волокна,
- неврологическое исследование.
Наиболее распространенные формы заболевания могут быть диагностированы путем анализа ДНК из крови пациента.
При диагностике необходимо тесное сотрудничество невролога, генетика, реабилитолога, хирурга-ортопеда и протезиста. В соответствии с выводами обследования, даются рекомендации относительно индивидуального плана реабилитации, при необходимости, назначается ортопедическая операция.
Значительная изменчивость клинических признаков болезни, наряду с отсутствием знаний о ней у многих врачей, часто приводит к неправильной диагностике.
Лечение
Сегодня неизвестно причинно-следственное лечение Шарко-Мари-Тута. Единственный вариант – симптоматическая терапия, при которой очень эффективный междисциплинарный подход к заболеванию.
Важная часть программы лечения болезни Шарко-Мари-Тутта – санаторно-курортная терапия. Другие методы включают:
- ЛФК (разрабатывает атрофированные мышцы),
- использование ортопедических и протезных приспособлений, в частности, стелек для обуви, фиксаторов малоберцовой слабости,
- ношение устойчивой ортопедической обуви.
При необходимости проводится хирургическое лечение деформаций – контрактуры ахиллова сухожилия, коррекция свода стопы, стабилизация голеностопного сустава, устранение сколиоза.
Перспективы в лечении
Перспективы для лечения Шарко-Мари-Тута представили результаты некоторых исследований на моделях болезни у животных. До настоящего времени сообщалось о положительном эффекте миелинизации периферических нервов с ШМТ 1A у мышей. Эффективность показал витамин С, антагонисты прогестерона – онапристон и нейротрофин-3. Некоторые из этих веществ уже проходят клинические испытания. Когда можно будет лечить заболевание с помощью новейших методов, зависит от того, насколько быстро или медленно будут проводиться исследования.

Нейротоксичные вещества
Эти вещества токсичны для периферической нервной системы. Они могут вызвать ухудшение симптомов у пациентов с болезнью Шарко-Мари-Тута: адриамицин, алкоголь, амиодарон, хлорамфеникол, цисплатин, дапсон, дифенилгидантоин, дисульфирам, этионамид, глютетимид, гидралазин, изониазид, большие дозы витамина А, D, В6, метронидазол, нитрофурантоин, N2O (повторное вдыхание), пенициллин (только в высоких дозах, на границе интоксикации), пенициламин, пергексилин, таксол, винсистин, золото.
Литий, Мизомидазол и Золофт следует использовать с осторожностью.
Прежде чем принимать какое-либо лекарство, поговорите с врачом о его возможных побочных эффектах.
Итоги
Болезнь Шарко-Мари-Тута – это наследственная нейропатия. Она широко распространена во всем мире, встречается во всех расах и этнических группах. Хотя заболевание было обнаружено 3-мя врачами (Жан-Мари Шарко, Пьер Мари и Говард Генри Тут) в 1886 г, причины некоторых форм заболеваний остаются неясными.
Молекулярная генетика на сегодняшний день идентифицировала причинные мутации в 40 генах, внесла значительный вклад в молекулярно-генетическую классификацию БШМТ, в понимание патогенеза.
Наследование включает в себя все типы моногенной передачи – утосомно-доминантный, утосомно-рецессивный, гоносомно-доминантный.
Поскольку нет причинно-следственной терапии, текущая стратегия лечения основана на реабилитации, протезировании и ортопедическом лечении. Прогноз для пациентов при подавляющем большинстве форм благоприятный, поскольку он не снижает нормальную продолжительность жизни, хотя существенно влияет на ее качество.
 (Пока оценок нет)
(Пока оценок нет)